|
Aunque conocido desde hace varias décadas, el receptor estrogénico ha sido recientemente objeto de una nueva atención al introducirse en la clínica una nueva familia de fármacos denominados "moduladores selectivos del receptor estrogénico" cuyo valor terapéutico en el tratamiento de la osteoporosis y otras complicaciones de la menopausia ha superado claramente a los estrógenos naturales o sintéticos que son simplemente agonistas o antagonistas de este receptor. El receptor estrogénico es un factor nuclear estimulado por ligandos, responsable de los efectos biológicos de los esteroides hormonales estrogénicos. El receptor estrogénico pertenece a la superfamilia de receptores nucleares que incluyen los receptores esteroídicos, los receptores retinoicos, los PPARS y otros muchos aún no identificados. Todos estos receptores nucleares muestran un alto grado de homología y son activados por moléculas pequeñas, de carácter lipófilo, que se unen a los mismos en un área hidrofóbica específica para cada ligando. Se conocen tres tipos de receptores estrogénicos, las formas ERa, ERb y ERg (o ERb2), siendo los dos primeros los más estudiados. El ERa, clonado de células MCF-7 procedentes de un tumor de mama humano, es una proteína que contiene 595 aminoácidos y un peso molecular de 66 kDa. Está codificado por un gen localizado en el cromosoma 6. El ERb, fue clonado a partir de células de próstata de rata y un peso molecular de 53 kDa. El gen que expresa el ERb humano se localiza en el cromosoma 14, expresándose en una amplia variedad de tejidos, incluyendo los cardiovasculares, los riñones, los pulmones, el cerebro y el sistema nervioso central. Las diferencias estructurales entre ambos tipos de receptores hace posible el diseñar ligandos que se unan selectivamente a uno u otro, constituyendo la base molecular del concepto de los moduladores selectivos del receptor estrogénico (SERMs) Funcionalmente, los receptores ERa y ERb pueden ser divididos en 5 regiones (*). Ambos muestran una elevada afinidad hacia los estrógenos y una considerable homología en la región donde se unen al DNA. Desde el punto de vista farmacológico, el dominio de unión de ligandos, que muestra una homología del 60%, es el que más interesa ya que es la zona del receptor donde se fijarán de forma más o menos selectiva y con mayor o menor afinidad las diferentes moléculas ya sean estrógenos puros o moduladores selectivos. |
||
 |
La estructura del dominio de unión de ligandos de ambas isoformas del receptor ER ha sido totalmente dilucidada, lo que permite explicar las interacciones que tienen lugar entre agonistas, agonistas parciales, moduladores selectivos y antiestrógenos esteroídicos (antagonistas puros) y, lo que es más importante, diseñar nuevos fármacos que actúen selectivamente. Aunque los detalles conformacionales del dominio de unión de ligandos (que han sido determinados mediante análisis cristalográficos de los receptores con sus ligandos) superan el objetivo de esta revisión, diremos que este dominio está formado por 12 hélices situadas en tres capas antiparalelas y que la orientación de la hélice 12 (*) es crucial para que se unan uno u otro ligando. Además, los avances realizados en los últimos años en la dilucidación del mecanismo de acción de los receptores estrogénicos han permite descubrir inhibidores del receptor estrogénico que NO se unen al mismo en el dominio de unión de ligandos. En efecto, los diferentes estados conformacionales adoptados por el ER en presencia de diferentes ligandos influyen directamente sobre la actividad transcripcional regulando la unión a esta parte de la molécula de cofactores e indirectamente modulando la estabilidad del receptor. Se han identificado péptidos capaces de actuar sobre las interacciones receptor-cofactor, capaces de inhibir la actividad transcripcional del receptor. Esta posibilidad de interaccionar fuera del dominio de unión de ligandos también explicaría la resistencia al tamoxifeno que se observa después de un tratamiento prolongado. En este caso, se formaría en la región del dominio de unión de ligandos un nueva cavidad que permitiría el reclutamiento de los cofactores, incluso estando fijado el tamoxifeno al receptor, lo que hace que el antiestrógeno se transforme en estrógeno en lo que respecta a la respuesta celular. La importancia de este descubrimiento es obvia, ya que permite el diseño de fármacos que eviten la formación de esta cavidad de resistencia en la superficie del receptor. La acumulación de conocimientos sobre la estructura y función de los receptores estrogénicos conseguida en los últimos años, ha permitido el diseño de cientos de compuestos que interfieren con ellos. Algunos de los más importantes, varios de ellos ya conocidos, son Tamoxifeno: fue el primer antiestrógeno inicialmente diseñado como contraceptivo, pero es utilizado en la actualidad en el tratamiento del cáncer de mama. Pertenece a la familia de los antiestrógenos derivados del trifeniletileno al igual que el toremifeno, clomifeno, idoxifeno y droloxifeno. Raloxifeno: pertenece a la familia de los benzotiofenos y es el primer modulador selectivo de los receptores estrogénicos, utilizándose para el tratamiento de la osteoporosis postmenopaúsica. Presenta la ventaja de no unirse a los receptores estrogénicos del útero. Varios homólogos del raloxifeno están en curso de evaluación. |
|
|
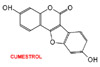
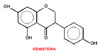
Fitoestrógenos: algunos flavonoides y benzopiranos ha sido extensamente estudiados como moduladores selectivos del receptor estrogénico. El cumestrol (*), un fitoestrógeno presente en el trébol y la alfalfa, tiene una propiedades parecidas a las del estradiol, mientras que la genisteína (*) se asemeja más al raloxifeno. Varios homólogos de la genisteína se encuentran en fase de investigación. Antagonistas estrogénicos puros: se han desarrollado algunos homólogos del estradiol, con estructura esteroídica como antiestrógenos puros, es decir con actividad antagonista estrogénica solamente. Estos compuestos, de los cuales el más significativo es el fulvestrant, son agentes antiproliferativos efectivos antagonizando el estradiol en los tumores de mama, pero no previenen la osteoporosis ni tienen ningún efecto sobre los lípidos. El fulvestrant se utiliza clínicamente en el tratamiento de los tumores de mama resistentes al tamoxifeno y en los tumores de mama y de útero metastásicos. |
|
REFERENCIAS
|
||
| Monografía creada : 26 de agosto de 2004. Equipo de Redacción de IQB |