|
||
|
SÍNDROME
DE BEHÇET [CD-10: M35.2]
|
|
|
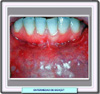
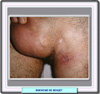
En 1937, el médico turco Dr. Hulusi Behçet describió un síndrome caracterizado por una triada de signos: estomatitis aftosa, úlceras genitales y uveitis (presencia de células inflamatorias en la cámara anterior del ojo). Las aftas orales recidivantes (100% de los casos) son dolorosas, y tienen un tamaño y aspecto variable curándose solas en 1 a 3 semanas, sin dejar cicatrices. Las aftas genitales (60-80%) se localizan en el glande y escroto en el varón y en la vulva, vagina y cervix en la mujer, siendo dolorosas y tardando en cicatrizar en el varón, y mucho menos molestas en las mujeres. La uveitis, bilateral casi siempre está presente en el 60-70% de los pacientes. A veces, se asocian coroiditis, hemorragias del cuerpo vítreo, neuritis óptica, alteraciones vasculares retinianas y otras que pueden conducir a la ceguera si no se trata la enfermedad. Los pacientes suelen tener más de 20 años y la enfermedad es unas dos veces más frecuente en el hombre que en la mujer. Sin embargo se han descrito casos en recién nacidos de madres con la enfermedad. La enfermedad neonatal está caracterizada por estomatitis aftosa y fenómenos sobre la piel que desaparecen espontáneamente a los 6 meses. La transmisión de la enfermedad de madres a hijos se debe a anticuerpos específicos que llegan al feto a través de la placenta. Aunque el síndrome de Behçet sólo es diagnosticado raras veces en los niños debe ser considerado en el diagnóstico diferencial de cualquier desorden inflamatorio multisistémico. El cuadro clínico del síndrome de Behçet en el niño difiere del del adulto en la menor frecuencia de manifestaciones oculares y en que aparecen otras manifestaciones menos corrientes. Entre estas se incluyen neutropenia, esplenomegalia, síndrome de Budd-Chiari, infiltrados pulmonares y ruptura de aneurisma de la arteria pulmonar. Los sistemas implicados en la enfermedad de Behçet son:
ETIOLOGIA Y PATOGENESIS La etiología y patogénesis de este síndrome permanecen sin esclarecer. Se considera como una enfermedad autoinmune ya que el denominador común en muchos pacientes es la vasculitis. Se han encontrado anticuerpos a membranas de la mucosa oral y complejos inmunes en el 50% de los casos. El antígeno HLA-B85 (B51 y B52) es de 3 a 4 veces más frecuente entre los pacientes que entre los controles. La enfermedad de Behçet es rara, de evolución crónica y con capacidad para producir inflamación a nivel de los vasos sanguíneos del organismo, por lo que se considera una vasculitis sistémica. Aunque inicialmente se consideró una enfermedad propia de los países mediterráneos y Japón, actualmente se sabe que la enfermedad está esparcida por todo el mundo. La creciente descripción de casos en los países asiáticos hace que muchos autores relacionen su distribución con las rutas comerciales remotas y le den el nombre de enfermedad de la Ruta de la Seda. La prevalencia de la enfermedad en España puede acercarse a los 5 casos por 100.000 habitantes, mientras que en Japón es de 13 y en Turquía de hasta 80 casos por 100.000 habitantes. No hay ninguna causa conocida responsable de la aparición de la enfermedad. No es contagiosa, ni se transmite sexualmente. Los investigadores piensan que aparece en personas genéticamente predispuestas que se ven expuestas a algún agente externo medioambiental, probablemente una bacteria. Los pacientes con la enfermedad suelen tener defectos en el sistema inmunológico. La evolución de la enfermedad de Behçet suele ser intermitente, con períodos de remisión y exacerbación a lo largo de los años, con una tendencia progresiva hacia la remisión. Los síntomas pueden durar desde días a semanas, o pueden comportarse de forma crónica durante meses o años. Tienden a causar un gran malestar y pueden provocar niveles de incapacidad importante que interfieren con la calidad de vida. Se basa en la observación clínica a largo plazo. Las clasificaciones diagnósticas mas utilizadas se muestran en la tabla. El diagnóstico diferencial incluye la aftosis oral recidivante, síndrome de Reiter, enfermedad de Crohn, colitis ulcerosa, esclerosis múltiple, LES y tumores cerebrales entre otros TRATAMIENTO Como la causa de la enfermedad es desconocida, el tratamiento se hace de acuerdo a los síntomas individuales y al momento de su aparición. Los medicamentos están dirigidos a reducir la inflamación o bien a intentar regular el sistema inmunológico. Algunos de los medicamentos usados son:
|
|
|
REFERENCIAS
|
||
|
|
|
|
|
|
Monografía revisada el 25 de abril 2011. Equipo de redacción de IQB | |
 |
||
|
|