
|
SINDROME
DE HALLERVORDEN-SPATZ [ICD-10: E78.3]
|
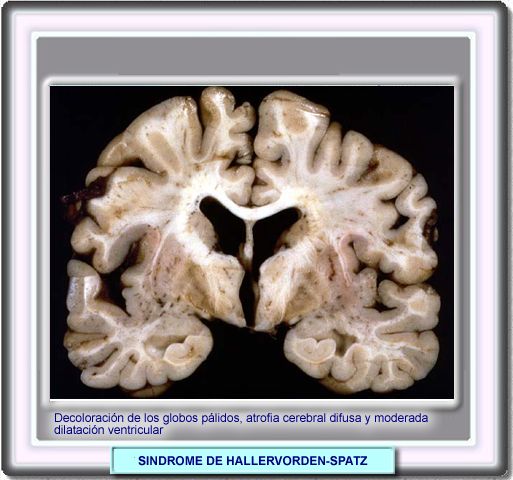 |
Este es un raro desorden neurológico caracterizado por signos piramidales, extrapiramidales y deterioro mental. La mayoría de los casos son familiares y el patrón de herencia corresponde a una transmisión recesiva autosómica, ocurre en todas las razas y tiene una frecuencia similar en ambos sexos. El inicio generalmente es en la infancia aunque se han descrito casos de comienzo en adultos. La mayoría tiene un curso fatal en 2 a 10 años. La forma clásica se inicia en la primera década de la vida con alteraciones corticoespinales (espasticidad, hiper-reflexia y reflejos plantares extensores). Aparecen signos de disfunción extrapiramidal que en ocasiones pueden retrasarse en varios años y usualmente ocurre en la forma de distonía. Ocasionalmente pueden observarse coreoatetosis y temblores y también se han descrito convulsiones La mayoría de los pacientes presentan retraso o deterioro intelectual y en algunos casos hay atrofia del nervio óptico, a menudo acompañada de retinitis pigmentosa. La enfermedad se debe a una mutación en el gen que codifica la pantotenato kinasa (PANK2) situado en el cromosoma 20p13-p12.3. Por este motivo, algunos autores proponen el nombre de degeneración neuronal asociada a la pantotenato kinasa para este enfermedad |


