ENFERMEDAD DE LA ORINA CON OLOR A JARABE DE ARCE [ICD-10: E71.0]
|
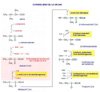
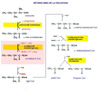
Una enfermedad debida a una deficiencia de las enzimas que metabolizan los aminoácidos ramificados valina, leucina e isoleucina. La acumulación de estos aminoácidos resulta en una encefalopatía y neurodegeneración que, si no se tratan adecuadamente, pueden conducir al coma y a la muerte. La valina, leucina e isoleucina son tres aminoácidos ramificados esenciales que son aportados por la dieta. Cuando la cantidad aportada excede de la necesaria para la síntesis de proteínas, son transaminados con a-cetoglutarato para formar a-cetoácidos ramificados que son oxidados mediante complejos enzimáticos muy similares a los que oxidan el piruvato y el a-cetoglutarato. La deficiencia de estos sistemas enzimáticos ocasiona la acumulación de estos productos en la sangre. DESCRIPCION La enfermedad con orina con olor a jarabe de arce se debe a mutaciones en al menos 4 genes (BCKDHA, BCKDHB, DBT y DLD), situados en el locus 19q13.1-q13.2, que codifican un complejo enzimático complejo de deshidrogenasas de a-cetoaminoácidos ramificados (BCKD - Branched-Chain alpha-Keto acid Dehydrogenase complex) que cataliza el catabolismo de la valina (*), leucina y isoleucina (*). El complejo BCKD, asociado a la membrana interna de la mitocondrial tiene tres componentes catalíticos (E1, E2, E3) y dos proteínas reguladoras, una BCKD-fosfatasa y una BCKD-kinasa. Además, el componente E1 tiene dos subunidades distintas llamadas E1a y E1b. La enfermedad muestra cuatro variantes según esté producida por alguno de los cuatro genes anteriores. Además, existen dos tipos adicionales causados por mutaciones de las proteínas reguladoras, una kinasa y una fosfatasa específicas. Las principales características clínicas de la enfermedad de la orina con olor a jarabe de arce son, además del olor de la orina característico a jarabe de arce (azúcar quemado) retrasos mental y del crecimiento. Existen cinco subtipos clínicos de la enfermedad, según estén afectados los cuatro genes antes señalados, o el gen E3 (que codifica tres sistemas enzimáticos mitocondriales: BCKD, alfa-cetoácido deshidrogenasa y piruvato deshidrogenasa):
La enfermedad se diagnostica a partir de los resultados de los análisis clínicos que muestran niveles plasmáticos elevados de aminoácidos ramificados. La presencia de aloisoleucina es definitivamente diaganóstica de esta enfermedad TRATAMIENTO El mejor tratamiento consiste en la restricción de aminoácidos ramificados en la dieta. En casos agudos en neonatos, se recurre a una infusión i.v. de glucosa e insulina para acelerar el anabolismo. Ocasionalmente, se puede recurrir a la diálisis para la eliminación de los aminoácidos ramificados. Se han descrito tres casos con éxito de transplantes hepáticos, si bien este tratamiento no se emplea con mayor frecuencia dado su elevado riesgo y al hecho de que la enfermedad se controla relativamente bien con una dieta restrictiva La forma que responde a la tiamina se trata con dosis de 10-20 mg de esta vitamina dos veces al día |
|
| Monografia creada: 11 de Julio de 2004. Equipo de Redacción de IQB | ||
| |