SINDROME DEL CROMOSOMA X FRÁGIL [ICD-10: Q99.2] |
|
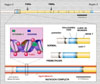
El síndrome de cromosoma X frágil (a veces llamado síndrome de Martin-Bell) es la causa más frecuente del retraso mental hereditario, ocurriendo en uno de cada 1.200 varones y una de cada 2.500 mujeres. Usualmente, los varones con este síndrome muestran retraso mental, facies dismórfica y macroorquidismo (*). Las mujeres muestran un fenotipo menos acentuado, siendo frecuente el fallo ovárico prematuro. Los individuos afectos de este síndrome muestran una fragilidad citogenética en la banda Xq27.3 del cromosoma X cuando las células se exponen a condiciones de cultivo pobres en ácido fólico. Dicho punto frágil se denomina FRAXA (retraso mental ligado al cromosoma A frágil tipo A). En 1991 se identificó el gen del síndrome del X frágil, el FMR1 que mostró contener la secuencia del trinucleótido CGG muchas veces repetida cerca del extremo 5' del gen. (*) Las mutaciones responsables del síndrome X implican la expansión de este segmento de repeticiones. Como regla general se aceptan los siguientes alelos para el FMR1:
|
|
|
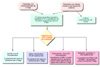
El diagnóstico se lleva a cabo mediante análisis del DNA. Analizando el tamaño del segmento formado por trinucleótidos CGG y el estado de metilación del gen FMR1, se puede determinar el genotipo tanto de los individuos afectados como de los portadores Se recomienda realizar un diagnóstico citogenético en (*):
Los signos del síndrome del X frágil en la primera infancia son inespecíficos siendo la manifestación usual un retraso en el desarrollo. Cualquier niño que se retrase en comenzar a hablar o a andar y con un desarrollo motor menor debe ser considerado candidato a un análisis citogenético, en particular si existente antecedentes familiares de retraso mental. Las condiciones que se deben considerar en el diagnóstico diferencial son el síndrome de Sotos, el síndrome de Prader-Willi, el autismo y el síndrome del déficit de atención-hiperactividad. Síndrome del XE frágil tipo E (FRAXE). Los individuos en el síndrome del XE frágil (fragilidad cromosómica en la banda q2.8 en los cultivos en ausencia de foltado, igualmente debida a la presencia de un exceso de trinucléotidos CGG en el gen FMR2) muestran un retraso mental menor que los del tipo A. A diferencia del síndrome de X frágil tipo A, el número de repeticiones puede sufrir una reducción o expansión a través de las generaciones. La reducción se produce cuando la transmisión es de padres a hijas y la expansión cuando pasa de madres a hijos, por lo que a veces se observan saltos generacionales. El síndrome del X frágil no tiene tratamiento. Se utilizarán las medidas adecuadas para el tratamiento de las alteraciones del comportamiento. Si hay fallo ovárico prematuro, se administrará terapia hormonal sustitutiva
|
|
REFERENCIAS
|
||
|
||
Monografía revisada el 7 de Enero de 2007. Equipo de Redacción de IQB
|
||
|
|