|
|
||
|
|
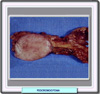
INTRODUCCION Los feocromocitomas son tumores relativamente raros que se originan en las células cromafines de la médula adrenal. Los feocromocitonas excretan catecolaminas y, por consiguiente ocasionan episodios intermitentes o sostenidos de hipertensión. La hipertensión producida por este tipo de tumores desaparece cuando el tumor es eliminado. También pueden aumentar la producción de otras hormonas suprarrenales como el ACTH, la somatostina y otros péptidos, por lo que ocasionalmente se pueden encontrar asociados a otros síndromes como el de Cushing. Los feocromocitomas pueden ser malignos, siendo importante el diagnóstico precoz y el tratamiento para mejorar el pronóstico. Según las diversas fuentes, la malignidad de los feocromocitomas oscila entre el 5% y el 46%. Pueden estar asociados a otros desórdenes endocrinos y no endocrinos y los feocromocitomas bilaterales son componentes del síndrome de neoplasia endocrina múltiple (MEN) de tipo IIa y IIb. Los feocromocitomas son tumores de las células cromafines que se localizan en su mayor parte en la medula adrenal. Sin embargo, pueden encontrarse células cromafines en otras partes, en particular en el órgano de Zuckerkandl. Cuando son extra-adrenales la mayor parte de los feocromocitomas son malignos. Los datos existentes sobre series de pacientes con feocromocitoma esporádico muestran que la adrenal derecha es más frecuentemente afectada que la izquierda. El tamaño del tumor oscila entre 3 y 5 cm (*), suelen ser de color gris y de consistencia blanda (*) . Microscópicamente se parecen a las células de las que proceden, estando las células tumorales separadas de la corteza adrenal por bandas de tejido fibroso (*) La distinción patológica entre los feocromocitomas benignos y malignos no está muy clara. En varias series de pacientes la recurrencia ha sido del 10 al 46% lo que indicaría un porcentaje de malignidad relativamente alto. Los tumores malignos suelen ser mayores y de más peso pero el único criterio absoluto de malignidad es la presencia de tumores secundarios en sitios donde no suele haber células de cromafina. Curiosamente, los feocromocitomas benignos suelen mostrar un pleomorfismo nuclear más acentuado que los malignos. Los feocromocitomad malignos suelen mostrar más mitosis que los benignos pero la invasión capsular y vascular es similar para ambos. El análisis del ploidismo del DNA muestra un DNA diploide en los feocromocitomas benignos mientras que en los malignos puede ser aneuploide, tetraploide e incluso poliploide. También se ha observado un mayor expresión del neuropéptido Y en los tumores benignos (9 de 9 casos frente a 4/11 en el caso de feocromocitomas malignos) MANIFESTACIONES CLINICAS Los pacientes con feocromocitoma presentan una serie de síntomas que van desde una hipertensión lábil moderada, hasta la muerte súbita por crisis hipertensiva, infarto de miocardio o accidente vascular cerebral. El paciente más clásico muestra cefaleas paroxísticas, palpitaciones, hipertensión y diaforesis. En el 50% de los casos la hipertensión es sostenida, mientras que es intermitente en el resto. En la mayor parte de los niños con feocromocitoma, la hipertensión es continua. Por regla general, existe un pérdida de peso, pero la obesidad no excluye el feocromocitoma. Son frecuentes los síntomas y signos de un aumento del metabolismo tales como aumento de la sudoración. La hipotensión ortostática es una consecuencia de la reducción de volumen plasmático debido al exceso de estimulación a-adrenérgica. Más o menos la mitad de los pacientes presentan intolerancia a los carbohidratos debido a la supresión de la insulina y a la estimulación de la glucosa hepática. Sin embargo, raras veces se requiere insulina y la intolerancia a los hidratos de carbono desaparece tan pronto el tumor es eliminado. |
|
|
|
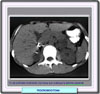
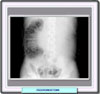
El diagóstico del feocromocitoma se basa en la medida de las catecolaminas y sus metabolitos en la orina. Si se sospecha clínicamente un feocromocitoma o el paciente tiene historia familiar de feocromocitoma o neoplasia endocrina múltiple de tipo IIa o IIb, la mejor prueba es realizar un análisis de 24 horas de catecolaminas, metanefrina y VMA (ácido vanilmandélico). También se debe determinar la creatinina para comprobar la significancia de la muestra de orina, y procurar en la medida de lo posible que el paciente no se encuentre medicado ni haya sido recientemente expuesto a medios de contraste radiográficos Catecolaminas libres: los valores normales de las catecolaminas oscilan entre 100 y 150 mg en 24 h. En la mayor parte de los pacientes con feocromocitoma se obtienen valores por encima de 250 mg/dia. Los valores de la epinefrina son valiosos ya que un esceso de esta sustancia (< 50 mg/día) se debe por regla general a una lesión adrenal. Se pueden obtener falsos positivos si el paciente se encuentra bajo medicación con metildopa, levodopa, atenolol y aminas simpaticomiméticas. Metanefrina y ácido vanilmandélico: en la mayor parte de los laboratorios se consideran valores normales de hasta 1.3 mg de metanefrinas totales y hasta 7 mg de VMA en 24 horas. Estos valores están considerablemente aumentados (más de tres veces el valor normal) en los pacientes con feocromocitoma Tanto la tomografía (*) como la resonancia magnética (*) permiten usualmente la visualización de los feocromocitomas. Radiológicamente también son visibles estos tumores (*)
|
|
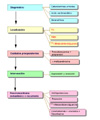 |
El tratamiento de elección (*) es la resección quirúrgica del tumor, lo que hace desaparecer usualmente la hipertensión. Antes de la operación es necesario controlar la presión arterial, utilizándose un a-bloqueante como la fenoxibenzamida y, si este no produce un efecto adecuado, con b-bloqueantes. La medicación se debe discontinuar la mañana de la cirugía. Administrar cloruro sódico isotónico como expansor del plasma. La mortalidad
es menor del 2% cuando intervienen un cirujano y un anestesista experimentados.
Actualmente, se prefieren los métodos laparoscópicos cuando
los tumores son inferiores a 8 cm. |
|
|
REFERENCIAS
|
||
| Monografía revisada 17 de Mayo de 2015. Equipo de Redacción de IQB | ||
|
|