COLÁGENO |
|
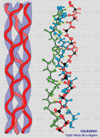
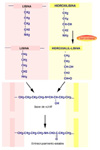
Se define una proteína como colágeno si contiene la triple hélice de colágeno de forma mayoritaria en su estructura molecular formando un agregado extracelular con una función predominantemente estructural. El colágeno es una proteína fibrosa insoluble que se caracteriza por contener grandes cantidades de una estructura regular formando un cilindro de una gran longitud. El colágeno se encuentra en todos los tejidos en los que sirve de armazón de sostén. Su importancia se corresponde con su elevado porcentaje: por ejemplo, supone el 4% del hígado, el 10% de los pulmones, el 50% del cartílago y el 70% de la piel. El colágeno está compuesto por tres cadenas que forman una triple hélice (*) Cada cadena tiene unos 1400 aminoácidos de los cuales uno de cada tres es una glicina. A intervalos regulares se encuentran otros aminoácidos, la prolina y la hidroxiprolina, poco frecuentes en otras proteínas. La presencia de estos aminoácidos particulares permite que las tres cadenas se enrrollen una alrededor de la otra formando una fibra muy resistente. Además, entre las cadenas se establecen puentes de hidrógeno que confieren al colágeno una gran estabilidad. Se conocen al menos 12 tipos de colágeno, numerados del I al XII (*). El más sencillo, de tipo I contiene una larga hebra de triple hélice que termina en los llamados telopéptidos (cada uno de los cuales finaliza en un -COOH o -NH2 terminal), que son pequeños segmentos que ya no tienen estructura superhelicoidal. Las moléculas de colágeno de tipo I se asocian una al lado de la otra mediante una reacción catalizada por una enzima específica la lisil-oxidasa que une la hidroxiprolina de una cadena con un resto de lisina de otra cadena. De esta manera se forman largas fibras. En otros tipos de colágeno, las cadenas finalizan por estructuras más o menos globulares. Por ejemplo, la membrana basal que soporta la piel está formada un colágeno de tipo IV (*) que tiene un extremo o cabeza globular y una cola extra. En la membrana basal, las cabezas se unen una con otras mientras que las colas se asocian de cuatro en cuatro formando unos complejos en forma de X. De esta manera se forma un retículo en el que otras moléculas (en este caso laminina y otros proteoglicanos) se entrecruzan formando una densa lámina (*) . El colágeno de tipo I está codificado por los genes COL1A1 y COL1A2. El colágeno de tipo II (*), abundante en el cartílago hialino, en el humor vítreo del ojo y en el núcleo pulposos de los discos intervertebrales está formado por fibras mucho mas gruesas. El colágeno de tipo III, está codificado por el COL1A3. Se encuentra en casi todos los tejidos en los que aparece el tipo I siendo excepciones, los huesos, los tendones y la córnea. El colágeno de tipo VI (*) se encuentra en muchos tejidos, incluyendo la aorta, los tendones y la piel. Es producido por los fibroblastos. La síntesis del colágeno se inicia en el citoplasma formándose cadenas aisladas que son llevadas al retículo endoplásmico donde los residuos de lisina y de prolina son hidroxilados, mediante sendas enzimas que requieren Fe+3 y vitamina C como cofactores (*) . La hidroxilación de la prolina hace termoestable a la proteína, mientras que la hidroxilacion de la lisina permitirá el entrecruzamiento de varias triples hélices. En este punto, las glicosil-transferasas del retículo endoplásmico glicosilan algunos restos de hidroxilisina. La triple hélice es ensamblada entonces quedando los extremos como polipéptidos libres, que pueden plegarse para formar las estructuras globulares. Las triples helices son transportadas al aparato de Golgi donde son modificadas por sulfatación, fosforilándose algunas serinas. El procolágeno resultante terminado es excretado de la célula a través de vesículas secretoras. La conversión del procolágeno en colágeno tiene lugar extracelularmente. Los telopéptidos terminales son hidrolizados por proteasas específicas y las triples hélices se ensamblan en fibrillas, momento en el que pueden participar otras proteínas del tejido conjuntivo como la laminina. Algunos de los restos de hidroxilisina son convertidos a aldehidos reactivos por la lisil-oxidasa, aldehidos que reaccionan con otros restos de lisina o hidroxilisina para formar los entrecruzamientos. ENFERMEDADES DEL COLÁGENO Se conocen algunos los detalles de los defectos que pueden producirse en la síntesis del colágeno y en las enfermedades asociadas:
|
|
|
||
Kuivaniemi et al. (1997) han revisado los datos sobre las cerca de 278 diferentes mutaciones encontradas en los genes que codifican los colágenos tipos I, II, III, IX, X, y XI de 317 pacientes no relacionados. La mayor parte de las mutaciones (217; 78% del total) fueron de una sola base, bien por intercambio de un codón de un aminoácido crítico bien por un empalme anormal del RNA. Las mutaciones de estos 6 colágenos ocasionan un amplio espectro de enfermedades del hueso, cartílago y vasos sanguíneos, incluyendo la osteogenesis imperfecta, una amplia variedad de condrodisplasias, síndrome de Ehlers-Danlos tipos IV y VII y, ocasionalmente, tipos raros de osteoporosis, osteoartritis y aneurisma familiar.
|
||
|
|
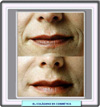
El colágeno se utiliza en cirugía cosmética en la llamada terapia de sustitución de colágeno, procedimiento que consiste en inyectar subcutáneamente colágeno natural o sintético en las áreas en las que se quiere que desaparezcan arrugas (*), cicatrices u otras imperfecciones de la piel. También se están utilizando como soporte en cultivos de células cartilaginosas para implantar posteriormente a pacientes que han sufrido lesiones, con una nueva tecnología denominada ingeniería de tejidos (*) |
|
REFERENCIAS
|
||
Monografía revisada el 30 Agosto de 2010
|
||
|
|