ENFERMEDAD DE ADDISON [ICD-10: E27.1] |
|
La enfermedad de Addison, también conocida como insuficiencia corticoadrenal primaria fue descrita por Thomas Addison como "un estado general de languidez y debilidad, desfallecimiento en la acción del corazón, irritabilidad del estómago y un cambio peculiar en el color de la piel". Los casos avanzados son relativamente fáciles de diagnosticar, pero el reconocimiento de los estadios más precoces suele ser bastante complicado. La enfermedad de Addison, poco frecuente, se debe a una destrucción autoinmune de la corteza suprarrenal lo que ocasiona una producción deficiente de glucocorticoides, mineralcorticoides y hormonas sexuales. La enfermedad de Addison se debe diferenciar de la insuficiencia corticoadrenal secundaria, con síntomas muy parecidos, en que esta se debe a una secreción deficiente de la ACTH, lo que a su vez reduce la secreción de los corticoides adrenales. En ambos casos, se trata pues, de una hipofunción de la corteza adrenal. La enfermedad de Addison es relativamente rara. Se estima una prevalencia de apróximadamente 110 casos/ millón de habitantes con una incidencia de 5 a 6 casos/millón/año.
|
|
|
La enfermedad de Addison resulta de la destrucción progresiva de las glándulas adrenales pudiendo llegar esta destrucción a ser de hasta el 90% antes de que aparezca una insuficiencia corticoadrenal clínicamente observable. Históricamente, la tuberculosis fue una causa frecuente de la enfermedad de Addison, aunque otras enfermedades como la histoplasmosis, coccidioimicosis y criptococosis también puede producir la enfermedad. Hoy día, la mayor parte de los casos de enfermedad de Addison se deben a una atrofia idiopática asociada a una enfermedad autoinmune . La enfermedad de Addison puede mostrarse aisladamente o como un componente de otras endocrinopatías, en particular hipo o hipertiroidismo, diabetes mellitus, tiroiditis linfocítica crónica, insuficiencia ovárica precoz, etc., constituyendo los síndrome autoinmunes poliglandulares (también denominados síndromes poliendocrinos de tipo autoinmune). La combinación de dos más de estas condiciones endocrinas autoinmunes constituye el síndrome autoinmune poliglandular de tipo 2, que puede ir acompañado de otras condiciones como la anemia perniciosa, vitiligo, alopecia, esprue no tropical y miastenia grave. El síndrome autoinmune poliglandular de tipo 2 se debe una mutación de un gen localizado en el cromosoma 6. La combinación
de una insuficiencia corticoadrenal y paratiroidea con una moniliasis
mucocutánea crónica constituye el síndrome autoinmune
poliglandular de tipo 1. Suele ir acompañado de otros desórdenes
entre los que se incluyen la anemia perniciosa, hepatitis, alopecia
e insuficiencia gonadal precoz. |
||
 |
Las tres
zonas de las glándulas adrenales son progresivamente destruidas
y sustituidas por tejido fibroso. La médula no es atacada pero
se muestra atrófica. Se produce en primer lugar la destrucción
de la zona glomerulosa seguida meses o años más tarde
de la disfunción de la zona fasciculata. Las hemorragias e infecciones
agudas de las glándulas adrenales suelen ir asociadas a una pérdida
repentina de funcionalidad adrenal. |
|
|
La destrucción gradual de las glándulas adrenales se caracteriza por el comienzo de fatiga, debilidad, anorexia, náuseas y vómitos, pérdida de peso, pigmentación de la piel y de las mucosas, hipotensión e hipoglucemia. Los síntomas más típicos de la enfermedad de Addison se muestran en la tabla, según sean consecuencia de la reducción de la actividad mineral corticoide, glucocorticoide o sexual. La astenia
es el síntoma más importante: puede ser esporádica,
pero a medida que la función adrenocortical se deteriora el paciente
se encuentra cada vez más fatigado e incluso debe guardar cama.
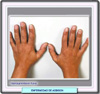
La hiperpigmentación (*)
puede ser muy importante o, por el contrario, no estar presente. Se
muestra como un color moreno o bronceado difuso que oscurece el codo,
los nudillos y otras partes ya pigmentadas como los pezones. En las
membranas mucosas pueden aparecer manchas azuladas. Algunos pacientes
desarrollan pecas y, al mismo tiempo, pueden presentarse áreas
de vitiligo (*). Es frecuente la presencia
de una hipotensión, con unos valores de 80/50 mmHg o menos. Los
síntomas de tipo gastrointestinal son variables y van desde un
ligera anorexia con pérdida de peso hasta una diarrea fulminante,
náuseas, vómitos, y dolor abdominal. Las mujeres suelen
perder vello axilar y púbico debido a la reducción de
los andrógenos |
|
|
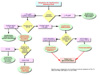
El diagnóstico de la insuficiencia adrenal se inicia con una sospecha basada en las manifestaciones clínicas, solo puede confirmarse las pruebas de estimulación del eje hipofisiario-pituitario-adrenal. Los síntomas y signos clínicos de la enfermedad de Addison se muestran en la tabla, habiéndose desarrollado un algoritmo de diagnóstico que permite evaluar la insuficiencia adrenocortical y diferenciar la insuficiencia primaria (Enfermedad de Addison) de la insuficiencia secundaria, basado en una serie de pruebas de estimulación del eje HPA Dado que
la debilidad y la fatiga son síntomas comunes a muchas enfermedades
el diagnóstico de la insuficiencia adrenocortical puede ser difícil.
Sin embargo, la combinación de efectos gastrointestinales, pérdida
de peso, anorexia e hiperpigmentación puede hacer sospechar una
insuficiencia adrenocortical y obligar a la realización de una
prueba de estimulación del eje hipotálamo-pituitario-adrenal
(HPA). |
|
|
Todos
los pacientes con insuficiencia corticoadrenal deben recibir un tratamiento
hormonal sustitutorio. Estos pacientes requieren una cuidadosa educación
para que puedan autocontrolarse, administrándose las dosis adecuadas
de corticoides. Para algunos autores, la hidrocortisona es el fármaco
de elección, siendo las dosis más usuales entre 20 y 30
mg al día según el peso del enfermo. El fármaco
se deberá administrar preferentemente con las comidas o con un
poco de leche o un antiácido, debido al efecto irritante que
tienen los corticoides sobre la mucosa gástrica. Otros autores
prefieren la dexametasona, En todos los casos, las dosis de los corticoides administrados se deben ajustar en función de la respuesta, que debe ser la normalización de los síntomas clínicos y de los niveles de ACTH por la mañana. Como la administración de hidrocortisona o dexametasona no sustituye la actividad mineralcorticoide de las hormonas adrenales, suele ser también necesaria la suplementación mineralcorticoide. La fluorhidrocortisona en dosis de 0.05 a 0.1 mg/día suele ser el fármaco empleado. Además, los pacientes deben asegurarse de una ingesta de sodio de entre 3 y 4 g/día. La adición de La respuesta a la medicación mineralcorticoide se determina a partir de medidas de la presión arterial y de los electrolitos, que deben mantenerse dentro de los límites de la normalidad. También puede ser útil la medida de la renina plasmática. En las mujeres con insuficiencia adrenal, los niveles de andrógenos suelen ser también bajos. Algunos médicos recomiendan la administración de 25 a 50 mg de DHEA (dihidroepidandrosterona) Crisis adrenal aguda: la insuficiencia adrenal aguda puede ser mortal. Tan pronto como el diagnóstico sea confirmado y/o se tengan los resultados de las pruebas. Debe iniciarse inmediatamente un tratamiento glucocorticoide empírico. La deficiencia mineralcorticoide se trata con suero salino y líquidos, no siendo necesaria aldosterona. Una vez estabilizado el paciente se prosigue el tratamiento con fluidos intravenosos durante 24 a 48 horas. Una vez confirmado el diagnóstico, la dexametasona es sustituída por hidrocortisona en dosis de 100 mg cada 8 horas. Estas dosis se ajustarán una vez resuelta la crisis hasta obtener las dosis de mantenimiento. Cuando el paciente se ha estabilizado, se iniciará el tratamiento mineralcorticoide con fluorhidrocortisona Situaciones
especiales: en
circunstancias especiales (estrés, cirugía, embarazo,
etc), los pacientes con insuficiencia adrenal crónica pueden
necesitar corticoides suplementarios. Algunas de las circunstancias
en las que puede ser necesaria un tratamiento especial, se muestran
en la tabla |
|
REFERENCIAS
|
||
|
||
|
||
|
||
|
||
| Monografía revisada el 14 de Mayo de 2014. Equipo de Redacción de IQB | ||
 |
||
| |