


|
|
|
DESCRIPCION La fenitoína es una hidantoína que se utiliza por vía oral y parenteral como anticonvulsivo. Se prescribe en el tratamiento profiláctico de las convulsiones tónico-clónicas (gran mal) y crisis parciales con sintomatología compleja (crisis psicomotoras). La fenitoína se puede utilizar como monofármaco o en combinación con otros fármacos anticonvulsivantes como el fenobarbital. También se puede utilizar para prevenir las convulsiones que se presentan durante la cirugía pero no se utiliza en las ausencias (pequeño mal). La fenitoína ha sido empleada en muchas otras condiciones incluyendo el tratamiento del dolor neuropático y arritmias cardíacas asociadas a un aumento del intervalo QT o producidas por el digital. El manejo clínico de la fenitoína es mas complicado que el de otros anticonvulsivantes debido a que posee una farmacocinética no lineal, se une en gran medida a las proteínas del plasma y muestra una alta variabilidad interindividual en su biodisponibilidad. Mecanismo de acción: los fármacos anticonvulsivantes elevan el umbral de las convulsiones y/o reducen la intensidad de la descarga. La fenitoína ejerce su efecto limitando la difusión de las descargas y su propagación a diferencia del fenobarbital y de la carbamazepina que elevan el umbral de las convulsiones. Por este motivo, la fenitoína es menos eficaz previniendo las convulsiones producidas por fármacos o las convulsiones electroinducidas. Los efectos de la fenitoína están relacionados con su acción sobre los canales de sodio de la membrana de la célula neuronal. La fenitoína ejerce sus efectos anticonvulsivantes con menos efectos sedantes que el fenobarbital. En grandes dosis, la fenitoína se muestra excitatoria e induce convulsiones. Por sus efectos sobre los canales de sodio, la fenitoína es ligeramente antiarrítmica, actuando sobre las fibras de Purkinje. Farmacocinética: la fenitoína se administra por vía oral y parenteral. En ambas formas, la fenitoína se puede presentar como ácido o como sal sódica, debiéndose tener en cuenta que la sal sódica contiene un 8% de fármaco inactivo, de modo que al pasar del uno al otro hay que reajustar la dosis. Si al pasar de la sal sódica al ácido no se redujera la dosis en un 8% podría producirse toxicidad. Las formulaciones de fenitoína son, por lo general, biodisponibles en un 90-100%, si bien la absorción varía según las diferentes formulaciones. En general la absorción es lenta debido a la baja solubilidad de la fenitoína. Las concentraciones máximas en el plasma se alcanzan a las 1.5 a 6 horas con las formulaciones "normales" mientras que en las formulaciones retardadas, el pico se alcanza a las 12 horas. Es importante tener en cuenta cuando se utiliza una suspensión de fenitoina que los alimentos reducen la biodisponibilidad de una forma significativa. La fenitoína se une extensamente a las proteínas del plasma (90-95%) aunque este porcentaje puede ser menor en los pacientes con hipoproteinemia o en los sujetos con insuficiencia renal. La fenitoína atraviesa la barrera hematoencefálica y se distribuye en el líquido cefalorraquídeo (LCR), la saliva, el semen, la bilis y los fluidos gastrointestinales. Las concentraciones del fármaco en el cerebro y en el LCR son idénticas a las concentraciones en sangre a los 10-20 minutos de una dosis intravenosa. El volumen de distribución de la fenitoína en condiciones de equilibrio ("steady state") es de 0.75 L/kg. Los neonatos y los prematuros muestran un volumen de distribución algo mayor. La fenitoína atraviesa la placenta y se excreta en la leche materna. En las mujeres embarazadas, las concentraciones fetales de fenitoína son idénticas a las de la madre. En los pacientes con insuficiencia hepática, al aumento de la bilirrubina en la sangre puede desplazar a la fenitoína de su unión a las proteínas, aumento la proporción de fármaco libre. En otras condiciones de hipoalbuminemia (grandes quemados, síndrome nefrótico, malnutrición, etc) también puede hacer un exceso de fenitoína libre. El metabolismo de la fenitoína presenta una elevada variabilidad. En efecto, este fármaco es uno de los pocos que pueden saturar la capacidad metabólica del hígado a concentraciones terapéuticas. Por debajo del punto de saturación, el metabolismo de la fenitoína es linear, siguiendo un proceso de primer orden. Sin embargo, cuando se alcanza o sobrepasa el punto de saturación, la eliminación de la fenitoína es mucho más lenta y tiene lugar mediante un proceso de orden cero. Por este motivo, la semi-vida de eliminación es la fenitoína es muy variable y puede oscilar entre las 7 y 42 horas para el mismo paciente, dependiendo de numerosos factores. Por otra parte, pequeños incrementos en las dosis pueden producir grandes elevaciones de los niveles plasmáticos si se alcanza el punto de saturación. Se recomienda, por tanto, la monitorización de los niveles plasmáticos. Cuando se
metaboliza, la fenitoína produce metabolitos inactivos. El aclaramiento
de la fenitoína es bajo y, además, está influenciada
por varios factores. Así una fiebre de 40º C durante mas de
24 horas puede inducir las enzimas hepáticas acelerando el metabolismo
de la fenitoína y su eliminación. Por el contrario las enfermedades
hepáticas reducen el metabolismo del fármaco y su eliminación
o, paradójicamente si la cantidad de fármaco unida a las
proteínas del plasma se reduce, al aumentar las concentraciones
de fármaco libre puede aumentar su eliminación. Por vía
renal solo se elimina el 5% de la dosis administrada. |
||
|
INDICACIONES Y POSOLOGIA
Dosis intravenosa inicial:
Dosis de mantenimiento por vía oral o intravenosa:
En el caso
de situaciones de emergencia, utilizar la fenitoína intravenosa.
|
||
|
Administración intravenosa:
Administración oral e intravenosa:
Administración intravenosa:
Administración intravenosa:
Administración oral:
Administración oral:
|
||
|
Monitorización de la fenitoína: En todos los pacientes tratados con fenitoína se deben determinar las concentraciones plasmáticas, independientemente de la indicación. Los niveles de fenitoína libre pueden ser útiles en los pacientes con hipoalbuminemia o con niveles elevados del nitrógeno ureico. Se consideran como concentraciones terapéuticas las situadas entre 10ďż˝20 µg/ml (equivalentes a 1ďż˝2 µg/ml de fenitoína libre). En algunos pacientes las concentraciones óptimas pueden llegar a los 25 µg/ml y algunas pocas personas pueden ser controladas con concentraciones de 8ďż˝10 µg/ml. Se requieren varios días o varias semanas para llegar a un estado de equilibrio ("steady-state"). Las manifestaciones
de la toxicidad a la fenitoína son muy variables: en algunos pacientes
con 20 µg/ml puede aparecer nistagmo lateral, la ataxia con 30 µg/ml,
y la disartria y letargia pueden aparecer cuando los niveles del fármaco
son > 40 µg/ml. Sin embargo, en otros pacientes se han encontrado
concentraciones de hasta 50 µg/m sin ningún síntoma
de toxicidad. |
||
|
Pacientes con insuficiencia hepática: dado que la fenitoína es metaboliza por el hígado, los pacientes con insuficiencia hepática pueden experimentar toxicidad. Adicionalmente, en muchas enfermedades hepáticas, como por ejemplo la cirrosis, existe hipoalbuminemia con lo que la concentración de fenitoína libre es mayor. En estos pacientes, las dosis de fenitoína se deben ajustar de acuerdo con los resultados de la monitorización de los niveles plasmáticos y de la respuesta clínica Pacientes con insuficiencia renal: si el aclaramiento de creatinina es 10 ml/min o más no se requieren ajustes en las dosis. Si el CrCl < 10 ml/min pueden ser necesarios reajustes de las dosis en función de los niveles plasmáticos y de la respuesta clínica. Diálisis
intermitente, continua o peritoneal: la fenitoína no es eliminada
de forma significativa durante una sesión de diálisis o
diálisis continua, por lo que se seguirá la pauta recomendada
para los pacientes con CrCl < 10 ml/min. |
||
|
CONTRAINDICACIONES La fenitoína no es eficaz en las ausencias (pequeño mal). Si se presentan simultáneamente convulsiones tónico-clónicas (gran mal) y ausencias (pequeño mal), es necesario un tratamiento combinado. La fenitoína no es eficaz en las convulsiones debidas a una hipoglucemia u otras causas metabólicas como por ejemplo, la hiponatremia. La retirada abrupta de la fenitoína puede desencadenar crisis convulsivas o un estado epiléptico. Si por cualquier razón fuera necesario discontinuar o sustituir la fenitoína por otra medicación, la retirada debe hacerse gradualmente. Sin embargo, si se observa una reacción alérgica o de hipersensibilidad grave, puede ser necesaria la rápida sustitución de la fenitoina por otro fármaco, que no deberá pertenecer a la familia de las hidantoínas. Los pacientes deben ser advertidos sobre la posibilidad de una reacción alérgica o hematológica grave, caracterizada por fiebre, dolor de garganta, rash, ampollas, úlcera en la boca, formación de cardenales, petequias, hemorragias purpúreas, linfadenopatías, síntomas similares a los del lupus eritematoso, anorexia, náuseas o vómitos o ictericia. Si cualquiera de estos síntomas se presentase, incluso después de un tratamiento prolongado, el paciente debe contactar inmediatamente con el médico. Por otra parte, la fenitoína no debe prescribirse a pacientes con historia de enfermedades hematológicas o alérgicas a menos que los beneficios superen los posibles riesgos. Se estima que la sensibilidad cruzada entre la fenitoina y los barbitúricos, la carbamazepina y la primidona oscila entre el 30 y 80%, lo que quiere decir que muchos pacientes alérgicos a estos anticonvulsivantes también lo serán a la fenitoína. La fenitoína se debe utilizar con precaución en los pacientes con discrasias sanguíneas producidas por fármacos o por alguna enfermedad hematológica debido al aumento potencial de toxicidad hemática por parte de la fenitoína, Aunque no demasiado frecuentes, algunas de las reacciones adversas que puede producir la fenitoína son leucopenia, neutropenia o trombocitopenia o incluso algunas más graves como la agranulocitosis o la anemia aplásica. El riesgo de desarrollar estos desórdenes es 5 veces mayor en los pacientes tratados con anticonvulsivantes en comparación con la población en general. Se recomienda un estudio hematológico previo a cualquier tratamiento con fenitoína, con frecuentes controles a intervalos regulares. Se debe considerar la retirada de la fenitoína si se produce la supresión de la función de la médula ósea. La fenitoína se debe utiliza con precaución en los pacientes con enfermedades cardiovasculares tales como arritmias cardíacas, insuficiencia cardíaca congestiva o enfermedad coronaria debido a que los síntomas de estas enfermedades pueden ser exacerbados. La fenitoína está contraindicada en los pacientes con anomalías de la conducción. La fenitoína intravenosa puede ocasionar arritmias ventriculares en pacientes con alteraciones de la función cardíaca debido a sus efectos sobre el automatismo. Por lo tanto, la fenitoína está contraindicada en el síndrome de Adam-Stokes, en los bloqueos de segundo y tercer grado y en la bradicardia sinusal. Estos efectos cardíacos de la fenitoína parenteral se deben, al parecer, al propilenglicol que se utiliza como disolvente. Es importante que la velocidad de la infusión i.v. no supere los 50 mg/min en los adultos con objeto de minimizar estos efectos. La probabilidad de reacciones cardiacas es mayor en los pacientes ancianos o debilitados. En estos pacientes se recomienda que la velocidad de la infusión no supere los 25 mg/min. No se recomienda la administración intramuscular de le fenitoína, debido su errática absorción por esta vía. Además, el pH de 10-12 de la solución inyectable origina una fuerte irritación local y un intenso dolor, sin contar de que puede producirse una necrosis aséptica. La fenitoína se elimina experimentando un metabolismo hepático que, en algunos sujetos, puede ser muy lento debido a una deficiencia genética de las enzimas metabolizantes. En estos individuos son necesarios ajustes muy precisos de las dosis de fenitoína. De igual forma, en los pacientes con alguna enfermedad hepática, el metabolismo de la fenitoina es más lento, ocasionándose niveles plasmáticos más elevados de lo normal. Por otra parte, en estos pacientes (en particular en los pacientes con cirrosis) suele presentarse hipoalbuminemia, lo que agrava aún más al situación, por existir más fenitoina libre en la sangre. La fenitoina puede ocasionar visión borrosa, somnolencia, mareos y fatiga, reduciendo el estado de alerta del paciente. Se debe advertir de esta posibilidad a los pacientes que deban conducir o manejar maquinaria que requiera una atención especial. Las concentraciones plasmáticas de fenitoína superiores a las normales pueden producir alteraciones psíquicas tales como delirio o psicosis que pueden evolucionar a una encefalopatía o, en muy raras ocasiones, a una disfunción cerebelar irreversible. Por este motivo, al menor síntoma de toxicidad se deben monitorizar los niveles plasmáticos reduciendo las dosis. Si a pesar de ello, los síntomas persistiesen, se debe discontinuar el tratamiento. En los enfermos
con una insuficiencia renal que produzca uremia deben ser también
monitorizados cuidadosamente. Las altas concentraciones de urea en sangre
desplazan a la fenitoína de las proteínas a las que se une
aumentando los niveles de fenitoína libre y, por tanto, la correspondiente
toxicidad. Pueden ser necesarios reajustes de las dosis en estos pacientes
|
||
|
|
La fenitoína se clasifica dentro de la categoría D de riesgo en el embarazo. La fenitoína es un teratógeno conocido que produce una serie de malformaciones fetales (conocidas como síndrome hidantoínico fetal). También se han descrito varios casos de cánceres, incluyendo neuroblastomas, en niños cuya madres recibieron fenitoína durante el embarazo. A pesar de que es un hecho probado de que la fenitoína (y otros anticonvulsivantes) producen efectos teratogénicos, existe una controversia acerca de si estos fármacos deben ser o no usados durante el embarazo ya que las convulsiones son igualmente lesivas para los fetos. Si una mujer queda embarazada durante el tratamiento con fenitoína, debe ser advertida de los posibles riesgos. En cualquier caso, debe evitarse la administración de dos o más anticonvulsivantes ya que se sabe que el riesgo de malformaciones fetales es menor cuando las convulsiones se tratan en monoterapia. Es aconsejable la administración de ácido fólico antes de la concepción y durante el embarazo, en particular durante el primer trimestre. También es recomendable la monitorización de los niveles plasmáticos debido que existe una correlación entre las concentraciones de fenitoína en la sangre de la madre y la incidencia de malformaciones en el feto. Especialmente en el segundo y tercer trimestre del embarazo, pueden alterarse la unión de la fenitoína a las proteínas del plasma, su metabolismo y su aclaramiento siendo necesaria una monitorización adecuada. Se han observado
defectos neonatales de la coagulación en las primeras 24 horas
en niños nacidos de madres tratadas con fenitoína o/y fenobarbital,
efecto que se debe a una depleción de vitamina K en el feto. La
administración de esta vitamina a la madre antes del parto o al
recién nacido puede corregir este defecto. |
|
|
|
La Academia
Americana de Pediatría considera compatible el uso de la fenitoína
con la lactancia. En efecto, sólo muy raras ocasiones se ha detectado
en el lactante alguna reacción adversa que pudiera ser atribuida
a la fenitoína (se ha descrito un caso de metahemoglobinemia).
El riesgo para el lactante puede ser minimizado si las concentraciones
plasmáticas del fármaco en la madre se mantienen dentro
de los límites aceptados |
|
|
La fenitoína puede estimular la secreción del glucagón y alterar la secreción de insulina. Estos efectos combinados pueden producir hiperglucemia. Se han comunicado casos de hiperglucemia e incluso de casos de cetoacidosis diabética en pacientes tratados con fenitoína. Se recomienda vigilar estrechamente los pacientes con diabetes mellitus, monitorizando con frecuente sus niveles glucémicos También deben ser estrechamente vigilados los pacientes con hipotiroidismo. La fenitoína reduce los niveles plasmáticos de las hormonas tiroideas con el correspondiente aumento de la hormona estimulante del tiroides en los pacientes previamente estabilizados con tiroxina. Se ha asociado osteomalacia a los tratamientos con fenitoína debido, al parecer, a una interferencia de este fármaco con la vitamina D. Por lo tanto, puede producirse un riesgo de osteopenia u osteoporosis en pacientes tratados con fenitoina Se han descrito casos de síndrome de Stevens-Johnson en pacientes tratados con fenitoína que fueron irradiados en la cabeza. Se recomienda tomar las debidas precauciones La fenitoína
disminuye la sensibilidad de los receptores colinérgicos por lo
que puede agravar el estado de los pacientes con miastenia grave. |
||
 |
INTERACCIONES Son muy
numerosas las interacciones que pueden tener lugar con la fenitoína.
Este fármaco es un inductor de casi todas las isoenzimas microsomales
del citocromo hepático P450, en particular las CYP3A4, CYP2D6,
CYP1A2, CYP2C9 y CYP2C19. Por lo tanto, la fenitoína puede acelerar
el metabolismo de todas las medicaciones que experimentan un metabolismo
mediado por estas isoenzimas. Sin embargo, la susceptibilidad de un paciente
a estas interacciones enzimáticas depende de muchos factores (edad,
tabaquismo, afecciones hepáticas, etc) de manera que no siempre
es posible predecir el comportamiento de la fenitoína frente a
un fármaco determinado. Por otra parte la fenitoína es,
a su vez, metabolizada por las isoenzimas CYP2C9 y CYP2C19, de manera
que muchos fármacos pueden inhibir o estimular su metabolismo según
que sean inhibidores o inductores del mismo. La inhibición del
metabolismo reduce el aclaramiento de la fenitoína (con el riesgo
de un aumento de los niveles plasmáticos y el consiguiente aumento
de la toxicidad), mientras que la inducción del metabolismo acelera
la eliminación (con el correspondiente riesgo de fracaso terapéutico).
Para complicar aún más la situación, la fenitoína
es un fármaco que une extensamente a las proteínas del plasma
pudiendo ser desplazada de las mismas por otros fármacos que también
se ligan a las proteínas. El aumento de la fenitoína libre
puede estar unido a un aumento de los efectos adversos. |
|
|
El aclaramiento de la fenitoína puede ser reducido por los fármacos o productos naturales que inhiben las enzimas microsomales del hígado, en particular las isoenzimas CY2C9 y CYP2C19. Algunos de los fármacos que inhiben estas enzimas mediante la inhibición de la CYP2C9 son la amiodarona, el cloramfenicol, la cimetidina, el clopidogrel, el fluconazol, la fluoxetina, la fluvastatina, la fluvoxamina, la isoniazida, el metabolito activo M1 de la leflunomida, el ketoconazol, el metronidazol, el miconazol cuando se administra sistémicamente, el modafinilo, el omeprazol, la ranitidina, el ritonavir, la sertralina, las sulfonamidas, el trimetoprim, y el zafirlukast. Entre los inhibidores del CYP2C19 se encuentran el fluconazol, la fluoxetina, la fluvoxamina, la fluvastatina, la isonizada, el ketoconazol, el modafinilo, el omeprazol, la oxcarbazepina, la sertralina, la lidocaína y el topiramato. En los pacientes que reciban estos fármacos concomitantemente con la fenitoína puede que sea necesario un reajuste de las dosis. Obsérvese que por su parte la fenitoína puede incrementar el metabolismo de los fármacos anteriores Se han publicado algunos casos raros de una reducción del metabolismo de la fenitoína después de la administración de la vacuna de la gripe. Los fármacos o productos naturales que inducen el metabolismo hepático en particular los que inducen las isoenzimas CYP2C y CYP2C19 pueden acelerar el metabolismo y la eliminación de la fenitoína. Algunos de los fármacos que tienen este efecto son la rifabutina, la rifampina y la rifapentina. A su vez muchos fármacos son afectados por la hidantoína (que es un inductor hepático), con lo que pueden ver su eficacia reducida cuando se administran concomitantemente. Algunos de los fármacos que pueden experimentar esta pérdida de eficacia son los inhibidores de la proteasa anti-retrovírica, la atorvastatina, el bupropion, los glucósidos cardíacos, los corticosteroides, la delavirdina, la doxiciclina, la guanfacina, la quinidina, el tamoxifen, el toramifen y la simvastatina. Otros fármacos que posiblemente sean afectados por la fenitoína son el paracetamol, el alosetrón, el bexaroteno, los antagonistas del calcio, la cevimelina, el citalopram, la disopiramida, el donepezilo, el estazolam, la galantamina, la levobupivacaina, la levodopa, la lidocaína, el mebendazol, la mexiletina, la mifepristona, el montelukast, el praziquantel, las hormonas tiroideas, el sildenafilo y el zaleplon. Dependiendo de la situación clínica de cada paciente, estas interacciones pueden ser más o menos significativas y no siempre pueden resultar en un fracaso terapéutico. Los fármacos inductores de las enzimas hepáticas pueden acelerar el metabolismo de los contraceptivos hormonales. Se han comunicado casos de embarazos no deseados cuando mujeres bajo tratamiento estrogénico fueron tratadas con fenitoína. Por lo tanto se recomienda adoptar otro procedimiento anticonceptivo que se deberá mantener al menos un mes después de discontinuar el tratamiento con la fenitoína. Obsérvese que la retirada de los estrógenos puede obligar a una reducción de las dosis de fenitoína El carbón activo, a veces utilizado para combatir la flatulencia, reduce la absorción digestiva de muchos fármacos, incluyendo la fenitoína. De igual forma, la alimentación entérica puede reducir de forma significativa la absorción de la fenitoína (hasta un 80%). Este efecto es atribuido al hecho de que la alimentación entérica reduce el tiempo de residencia gastrointestinal de la fenitoína y también al hecho de que el fármaco puede formar complejos poco absorbibles con el calcio, el magnesio o algunas de las proteínas presentes en el alimento. Es posible que el mismo tubo de alimentación entérica adsorba parte del fármaco. Se recomienda en estos casos, administrar el fármaco 1-2 horas después de la alimentación entérica o retrasar esta 1 a 2 horas después de la dosis de fenitoína. Si la alimentación entérica es continua, la situación es más complicada por lo que a veces se recurre a la administración intravenosa del fármaco. Si esto no fuera posible, la fenitoina debe ser diluida en suero salino, lavando el tubo varias veces con suero salino o con agua, Además, deben monitorizarse los niveles plasmáticos de fenitoína ajustando las dosis hasta conseguir los efectos terapéuticos deseados. Otros factores que pueden reducir la absorción digestiva de la fenitoína son los antiácidos que contienen magnesio o aluminio y el carbonato cálcico. Las sales de calcio producen complejos con la fenitoina que no puede ser absorbidos. Igualmente, el sucralfato puede reducir la absorción oral de la fenitoína si ambos fármacos se administran al mismo tiempo. Estas interacciones pueden minimizarse dejando pasar un mínimo de 2 horas entre la administración de ambos tipos de fármacos. El colestipol aumenta ligeramente la velocidad de la absorción de la fenitoína, pero la biodisponibilidad global no es afectada. La fenitoína es un inductor hepático y puede acelerar el metabolismo de otros anticonvulsivantes, incluyendo el clonazepam, el diazepam, el felbamato, la etosuximida, la lamotrigina, la oxcarbazina, la tiagabina, la topiramida y la zonisamida. Por el contrario, la fenitoína puede reducir el metabolismo del fenobarbital. El caso de la primidona es algo más complejo: por un lado el metabolismo de la primidona es acelerado pero, por otro, dado que la primidona es metabolizada a fenobarbital, pueden aumentar las concentraciones de este último. Por su parte, algunos de estos anticonvulsivantes pueden interferir el metabolismo de la fenitoína. La carbamazepina no tiene ningún efecto sobre las concentraciones plasmáticas de fenitoína; el felbamato, la metosuximida y el topiramato aumentan las concentraciones plasmáticas de la fenitoína, y lo mismo ocurre con el valproato. El fenobarbital y los barbitúricos tienen un efecto variable. La gabapentina, lamotrigina, levetiracetam, tiagabina y zonisamida no parecen afectar las concentraciones de fenitoína. La tizanidina aumenta los niveles plasmáticos de fenitoína hasta en un 100% produciendo somnolencia, aunque se desconoce el mecanismo de esta interacción. En efecto los estudios in vitro con microsomas hepáticos humanos no han evidenciado un efecto de tizanidina sobre los fármacos que son metabolizados por las isoenzimas del citocromo P450 La fenitoína (y la fosfenitoína) pueden inducir las enzimas hepáticas del citocromo P450 reduciendo las concentraciones plasmáticas de ciclosporina, sirolimus o tacrolimus. Si se añade la fenitoína a un tratamiento inmunosupresor, se deberán monitorizar y ajustar los niveles plasmáticos de estos fármacos para evitar un fracaso terapéutico. De igual forma, si se discontinua a la fenitoina a pacientes inmunodeprimidos, las dosis de los fármacos inmunosupresores deberán ser reajustadas para evitar su toxicidad. El micofenolato mofetilo, un inmunosupresor, no es afectado por la fenitoína. Sin embargo, este fármaco reduce ligeramente la fracción de fenitoína unida a las proteínas del plasma aumentando las concentraciones de fenitoína libre. Las interacciones entre la fenitoína y la warfarina son muy complejas: en primer lugar, la fenitoína puede desplazar a la warfarina de las proteínas a las que se une, aumentando los niveles de warfarina en sangre y produciendo un rápido aumento del INR. En segundo lugar, la administración crónica de fenitoína puede reducir la efectividad de la warfarina al inducir su metabolismo. En tercer lugar puede producirse una inhibición mutua competitivo dado que la warfarina y la fenitoína son ambos sustratos para el citocromo P450. El dicumarol ocasiona unas interacciones parecidas a las de la warfarina. En ambos casos, se recomienda una frecuente monitorización de los niveles plasmáticos de la fenitoína y del INR. Si se discontinua el tratamiento anticoagulante, las dosis de fenitoína deberá ser reajustadas convenientemente. El tramadol puede rebajar el umbral convulsivo e interferir con los efectos terapéuticos de los anticonvulsivantes. No se recomienda la administración de este analgésico en pacientes bajo tratamiento anticonvulsivante. Adicionalmente, el metabolismo del tramadol es acelerado por la fenitoína reduciendo su capacidad analgésica. El tratamiento crónico con fenitoína puede antagonizar los efectos de los bloqueantes musculares no despolarizantes. Esta interacción hace que el comienzo del bloqueo neuromuscular sea más lento y que la duración del mismo sea menor. Los anticonvulsivantes como la fenitoína pueden reducir la actividad de la vitamina D y de sus análogos incrementando su metabolismo. El tratamiento crónico con fenitoína ha estado asociado con raquitismo y osteomalacia. Puede ser necesario un suplemento de vitamina D si el aporte de esta vitamina en la dieta no es suficiente. De igual forma, la acetazolamida puede contribuir a una osteomalacia en pacientes tratados con fenitoína al aumentar la excreción urinaria de calcio. En estos pacientes también puede ser necesario un suplemento de vitamina D Las dosis elevadas de salicilatos (p.ej >2000 mg/día) pueden desplazar a la fenitoína de las proteínas del plasma a las que se encuentra unida, originando unos niveles de fenitoína libre mas elevados, con el correspondiente riesgo de toxicidad. Lo mismo puede ocurrir en el caso del ibuprofen, pero solo cuando las dosis son más elevadas que las que usualmente se utilizan. Los pacientes estabilizados con dopamina en infusión i.v. pueden experimentar una súbita hipotensión e incluso una parada cardíaca si se administra fenitoína intravenosa. Se desconoce si la fenitoína oral puede ocasionar un efecto similar. En cualquier caso se deberán extremar las precauciones si de administra fenitoína a pacientes bajo dopamina. La fenitoína aumenta el riesgo de la hepatotoxicidad de los anestésicos generales enflurano, halotano o metoxiflurano Debido a sus efectos inductores de los enzimas hepáticos, la fenitoína reduce la biodisponibilidad del itraconazol (hasta en el 93%) y reduce su semi-vida plástica casi 10 veces. A su vez, el itraconazol aumenta las concentraciones de la fenitoína en un 10%, lo que puede ocasionar en algunos pacientes un efecto clínico significativo. Existen complejas interacciones entre la fenitoína y el ácido fólico (vitamina B9). Se ha especulado sobre un efecto del ácido fólico que aceleraría el metabolismo de la fenitoína con la correspondiente pérdida del control sobre las convulsiones. Por otra parte, la actividad de la fenitoína parece depender en parte de una depleción de ácido fólico o de una inhibición de la conversión del ácido fólico a un derivado fácilmente transportable al cerebro. La administración de ácido fólico a los animales de laboratorio produce convulsiones, especialmente si las dosis son elevadas. No se recomienda la administración de ácido fólico a menos de que exista una clara evidencia hematológica de deficiencia de esta vitamina. Tampoco es recomendable administrar ácido folínico (leucovorina) a pacientes tratados con fenitoína ni consumir alimentos ricos en ácido fólico. La fenitoína puede acelerar el metabolismo de algunos fármacos antineoplásicos lo que afectaría su eficacia. Se ha comunicado un aclaramiento más rápido del busulfan, placlitaxel y teniposido cuando se añadió a la quimioterapia un tratamiento anticonvulsivante. Por su parte, algunos de estos fármacos (bleomicina, carboplatino, carmustina cisplatino, doxorrubicina, alcaloides del vinca) pueden ocasionar concentraciones subterapéuticas de fenitoína. En algunos pacientes tratados con cisplatino, las dosis de fenitoína tuvieron que ser incrementadas en un 20-100% para conseguir el control de las convulsiones. Se han observado concentraciones plasmáticas elevadas de fenitoína cuando se ha utilizado concomitantemente 5-fluoruracilo o capecitabina y tegafur. El metotrexato se une parcialmente a las proteínas del plasma por lo que los fármacos que también lo hacen, como la fenitoína o la fosfenitoína, son capaces de desplazarlo produciendo unas concentraciones plasmáticas más altas de metotrexato libre y un aumento de la toxicidad de este fármaco. Esta toxicidad puede ser significativa incluso cuando el metotrexato se utiliza en dosis pequeñas como en las enfermedades reumáticas. Adicionalmente, el metotrexato puede inducir un aumento del aclaramiento de la fenitoína. Por lo tanto, se recomienda monitorizar los niveles plasmáticos de fenitoína para asegurarse de que se mantienen unas concentraciones anticonvulsivantes adecuadas y para limitar la toxicidad del metotrexato. Se han detectado interacciones farmacocinéticas en pacientes con gliomas malignos tratados con irinotecan y fenitoína (u otros anticonvulsivantes como la carbamazepina o el fenobarbital). Los anticonvulsivantes aumentan en un 200% el aclaramiento del irinotecan con la correspondiente reducción de la AUC de este fármaco. Además, cuando al irinotecan se añade dexametasona se observa un importante aumento de la excreción urinaria de la medicación antineoplásica lo que puede conducir a una pérdida de la eficacia terapéutica. La administración concomitante de alcaloides de la vinca con la fenitoína reduce en más de un 50% las concentraciones plasmáticas del anticonvulsivante con una perdida de su eficacia. Esta reducción comienza a observarse a las 24 horas y puede mantenerse 10 días o más después de la administración de la vincamina o de la vinblastina. Se ha comunicado un aumento de las concentraciones plasmáticas de fenitoína cuando se utiliza concomitantemente el levamisol. Este efecto se debe a que el levamisol reduce el metabolismo de la fenitoína La nilutamida inhibe la actividad del sistema enzimático dependiente del citocromo P450 y, por lo tanto reduce el metabolismo de todos los fármacos que son metabolizados por este, incluyendo la fenitoína. En consecuencia, puede ser necesario reducir las dosis de fenitoína si se administra concomitantemente la nilutamida. Los fármacos que inducen las isoenzimas CYP1A2 como la fenitoina aumentan el metabolismo oxidativo de las xantinas como la cafeína y las teofilinas. Es posible que sea necesario aumentar las dosis de teofilina en los asmáticos cuando se inicie un tratamiento con fenitoína. Pero, lo que es más grave, puede aparecer una seria toxicidad teofilínica si se discontinua la fenitoína y no se reduce adecuadamente la dosis de teofilina. Por otra parte, las xantinas pueden interferir con la absorción gastrointestinal de la fenitoína. Se han publicado pocos casos de toxicidad por litio asociada a la administración concomitante de fenitoína, y en los pocos casos conocidos, se desconoce cual es su mecanismo. Dado el estrecho margen terapéutico de ambos fármacos se recomienda una estrecha vigilancia de los pacientes con objeto de detectar cualquier síntoma de toxicidad inducida por litio. La amfetamina y la dextroamfetamina retrasan la absorción intestinal de la fenitoína desconociéndose si también afecta a su absorción. Por otra parte, las amfetaminas pueden rebajar el umbral convulsivo en algunos pacientes. Igualmente, la pemolina rebaja el umbral convulsivo reduciendo la eficacia de los anticonvulsivantes y haciendo necesarios reajustes en las dosis. Otros psicoestimulantes también pueden rebajar el umbral convulsivo si bien no existen comunicaciones que señalen problemas sobre el estado convulsivo de pacientes tratados con ambos tipos de fármacos Por su parte, el metilfenidato inhibe el metabolismo de la fenitoína incrementando los niveles plasmáticos. Puede ser necesaria una reducción de las dosis de la fenitoína cuando el metilfenidato se administra concomitantemente. El diazoxido puede aumentar el metabolismo hepático de la fenitoína, si bien se desconoce la significancia clínica de esta interacción. Se ha comunicado un caso en el que la administración de aciclovir redujo de forma significativa las concentraciones plasmáticas de fenitoína con la pérdida correspondiente del control sobre las convulsiones. Aunque no hay mas datos, los clínicos deben tener en cuenta que puede que sea necesario aumentar las dosis de fenitoína si se administra aciclovir (o valaciclovir) simultáneamente La fenitoína pueden presentar efectos depresores del sistema nervioso central que pueden adicionarse a los de los otros fármacos con la misma actividad como el alcohol, los ansiolíticos, los sedantes e hipnóticos y los antagonistas H1 sedantes que se utilizan como inductores del sueño La amitriptilina, la clomipramina y la imipramina son sustratos para las isoenzima CYP2C lo mismo que la fenitoína pudiendo producirse un estado competitivo que puede conducir a un aumento de las concentraciones plasmáticas de fenitoína. Por su parte, la fenitoína puede inducir las enzimas microsomales hepáticas aumentando el metabolismo y aclaramiento de los antidepresivos tricíclicos. Por otra parte, estos fármacos y otros antidepresivos similares (p.ej. la amoxapina y la maprotilina) rebajan el umbral convulsivo, lo que puede representar un problema en particular si se utilizan en dosis elevadas. Se recomienda la frecuente monitorización de los niveles plasmáticos de fenitoína si se utilizan concomitantemente antidepresivos tricíclicos Lo mismo puede ocurrir con los fármacos antipsicóticos (clozapina, haloperidol, loxapina, pimozida, quetiapina, risperidona, tioridazina, y las fenotiazinas) cuyo metabolismo es acelerado por la fenitoína. Adicionalmente las fenotiazinas y algunos otros antipsicóticos pueden aumentar la depresión del SNC y rebajar el umbral convulsivo. Es necesaria la monitorización de los niveles plasmáticos de la fenitoina y, probablemente se deberán aumentar la dosis para conseguir un adecuado control de las convulsiones. La trazodona interacciona de una manera idéntica a los antipsicóticos: por un lado su metabolismo es acelerado por la fenitoína y por otra parte, la trazodona rebaja el umbral convulsivo debido a sus efectos depresores del SNC. Finalmente, la trazodona produce una somnolencia que puede ser aditiva a la provocada por la fenitoína Algunos estudios sugieren que la paroxetina no afecta a las concentraciones plasmáticas de fenitoína. Sin embargo, se ha reportado un caso en el que la administración concomitante de la paroxetina durante 4 semanas originó un aumento de las concentraciones plasmáticas de fenitoína con los correspondiente síntomas de toxicidad. Se desconoce el motivo de esta elevación ya que la paroxetina es un inhibidor de la isoenzima CYP 2D6 que no interviene en el metabolismo de la fenitoína. En cualquier caso se recomienda precaución si se administran fenitoina y paroxetina simultáneamente Aunque no se ha confirmado de una forma definitiva parece ser que el interferón 1b podría afectar las concentraciones de enzimas del citocromo P450, incluyendo las que metabolizan la fenitoína. Hasta que se obtengan más informaciones acerca de esta interacción se recomienda precaución si se administra interferón a los pacientes tratados con fenitoína. Se publicado informes contradictorios acerca de la influencia de la ciprofloxacina sobre las concentraciones plasmáticas de la fenitoina. La ciprofloxacina es un inhibidor conocido de la isoenzima CYP 1A2 que no participa en el metabolismo de la fenitoína. Por lo tanto, hasta que se disponga de más datos acerca de esta posible interacción se recomienda precaución si se prescriben ciprofloxacina y fenitoína concomitantemente Se ha publicado un caso de un paciente epiléptico con psoriasis que fue tratado con metoxsaleno + luz UV, en el que la fenitoína indujo una reducción significativa de la AUC del metoxsaleno y a una pérdida del efecto terapéutico de este fármaco. Al discontinuar la fenitoína manteniendo la medicación antipsoriásica, el paciente desarrolló eritema y ampollas como consecuencia del aumento de los niveles plasmáticos del metoxsaleno El disulfiram puede interferir con el metabolismo de las hidantoínas, aumentando sus niveles plasmáticos y su toxicidad. Se recomienda la monitorización de los niveles plasmáticos de fenitoína antes y durante la administración del disulfiram, ajustando las dosis de ambos fármacos de acuerdo con los valores obtenidos. De igual forma, si el disulfiram es discontinuado es probable que sea necesario un reajuste de las dosis La ingestión crónica de alcohol induce las isoenzimas microsomales hepáticas y aumenta el aclaramiento de la fenitoína, Además, el alcohol posee un cierto potencial epileptogénico que puede anular el control de la epilepsia por algunos fármacos. Se debe evitar el consumo de alcohol por parte de los pacientes bajo fenitoína o carbamazepina, si bien la ingesta ocasional de alcohol en sujetos no alcohólicos no parece afectar al metabolismo hepático de la fenitoína. La administración concomitante de fenitoína con opiáceos puede hacer necesaria un reajuste de las dosis de estos para conseguir una analgesia adecuada o para prevenir el síndrome de abstinencia una vez discontinuados. Adicionalmente, los opiáceos tienen un efecto depresor sobre el sistema nervioso central, efecto que puede sumarse al de la fenitoína. Los pacientes
tratados con prilocaína y fenitoína presentan un riesgo
mayor de desarrollar una metahemoglobinemia |
||
|
|
El
zumo de pomelo contiene la 6,7-dihidroxibergamotina, una furanocumarina
que inhibe la CYP 3A4 de los enterocitos y, por lo tanto se recomienda que
este zumo de fruta no sea consumido durante un tratamiento con carbamazepina,
ya que puede producirse un aumento de la sedación y otros efectos
adversos. |
|
|
|
Las
sustancias que actúan sobre el sistema nervioso central, incluyendo
los agentes anticonvulsivantes, pueden interaccionar con el kava-kava. Esta
interacciones son de carácter farmacodinámico o pueden resultar
de un mecanismo aditivo. Las pacientes tratados con anticonvulsivantes deben
consultar con el médico el consumo de cualquier hierba o preparado
que contenga kava-kava |
|
|
|
La
hierba de San Juan (Hypericum perforatum) es un inductor de las
enzimas microsomales del citocromo P450 y puede acelerar el metabolismo
y eliminación de la fenitoína produciendo un fracaso terapéutico |
|
| La
fenitoína puede interferir en las siguientes pruebas de laboratorio:
metirapona, dexametasona, yodo unido a proteínas, glucosa, fosfatasa
alcalina y GGT. |
||
|
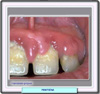
REACCIONES ADVERSAS Aunque la fenitoína es considerada como un fármaco potencialmente tóxico, siguiendo las pautas de dosificación y las instrucciones recomendadas, su administración es segura y eficaz sin que aparezcan generalmente efectos secundarios. Los principales signos de toxicidad asociados con la administración intravenosa de fenitoína son colapso cardiovascular y/o depresión del sistema nervioso central. Cuando se administra rápidamente puede aparecer hipotensión. Las posibles reacciones adversas se han clasificado por órganos-sistema:
|
||
 |
|
|
|
||
|
||
|
||
|
|
PRESENTACION
|
|
|
REFERENCIAS
|
||
|
||
Monografía
revisada el 16 de Julio de 2014.Equipo de redacci�n de IQB (Centro colaborador de La Administraci�n Nacional de Medicamentos, alimentos y Tecnolog�a M�dica -ANMAT - Argentina).  |
||
|
|


